09122010 chemoinformatics
これは購入しておく。
 Free Energy Calculations in Rational Drug Design
Free Energy Calculations in Rational Drug DesignSpringer US / ¥ 13,115 ()
一時的に在庫切れですが、商品が入荷次第配送します。配送予定日がわかり次第Eメールにてお知らせします。商品の代金は発送時に請求いたします。
Free energy calculations represent the most accurate computational method available for predicting enzyme inhibitor binding affinities. Advances in computer power in the 1990s enabled the practical application of these calculations in rationale drug design. This book represents the first comprehensive review of this growing area of research and covers the basic theory underlying the method, numerous state of the art strategies designed to improve throughput and dozen examples wherein free energy calculations were used to design and evaluate potential drug candidates.
うちの職場は古きよきというか(略)
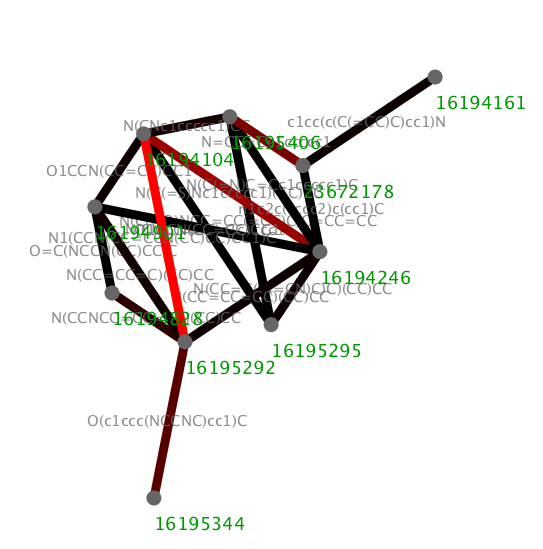
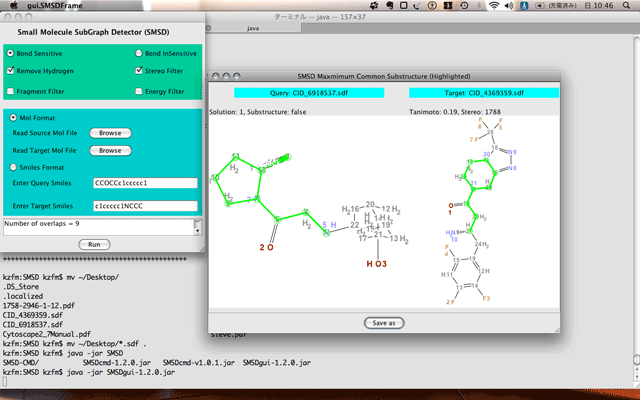
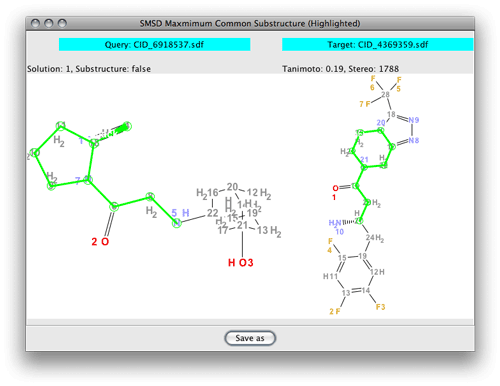
量子化学計算のコストも下がったというか、小さい化合物だったら普通に待てる時間でレスポンス返せるのでWebインターフェースつくってある。配座の安定性調べたり、ソルベーションエナジー計算したりとか色々やれるので、合成力(how to make)よりも、何を合成するか(what to make)?つまり、どういう仮定を置くか、論理をどう組み立てるか?という化学反応の背景にある物理化学とか量子化学に対する理解を強めるのは重要でないのかなぁと思っているんだけど。
どうかな?
 ケモインフォマティックス―予測と設計のための化学情報学
ケモインフォマティックス―予測と設計のための化学情報学 入門Mercurial Linux/Windows対応
入門Mercurial Linux/Windows対応
 Handbook of Chemoinformatics Algorithms (Chapman & Hall/Crc Mathematical and Computational Biology)
Handbook of Chemoinformatics Algorithms (Chapman & Hall/Crc Mathematical and Computational Biology) Chemoinformatics and Computational Chemical Biology (Methods in Molecular Biology)
Chemoinformatics and Computational Chemical Biology (Methods in Molecular Biology)