
22072025 chemoinformatics
GFlowNetやその他の手法を比較すると次のようになるらしい(ちょっと微妙に違うような気もするが)
| モデル | 多様性 | 解釈性 | サンプリング速度 | 合成容易性 |
|---|---|---|---|---|
| GFlowNet | ◎ | ◎ | ◎ | ○ |
| VAE / JT-VAE | △ | △ | ◎ | ◎ |
| GAN / Flow | △ | × | ○ | × |
| 強化学習 (REINVENTなど) | ○ | ○ | × | ○ |
| MCMC(伝統的) | ○ | ◎ | × | △ |
22072025 chemoinformatics
GFlowNetやその他の手法を比較すると次のようになるらしい(ちょっと微妙に違うような気もするが)
| モデル | 多様性 | 解釈性 | サンプリング速度 | 合成容易性 |
|---|---|---|---|---|
| GFlowNet | ◎ | ◎ | ◎ | ○ |
| VAE / JT-VAE | △ | △ | ◎ | ◎ |
| GAN / Flow | △ | × | ○ | × |
| 強化学習 (REINVENTなど) | ○ | ○ | × | ○ |
| MCMC(伝統的) | ○ | ◎ | × | △ |
21072025 chemoinformatics
3連休の後半はどこかに行こうと予定を考えていたのだけど、生憎風邪をひいてしまい(喉は痛いが熱はない)家で大人しくする羽目に。論文読んだりコード書いたりで捗るといえば捗ったので悪いことではないのかも。以下のスライドもよくまとまっていて面白かったです。
オフィシャルなリポジトリではapple silicon(mps)には対応していないのだけど、こちらであればmps使えるのでインストールして動かしてみた。
ちょっと興味があったのがchignolinの8番目の残基を変えるとターンの構造が変わるっていう論文で、そういうの再現するのかねーと。
コマンドはこんな感じでOK
boltz predict --use_msa_server sig2.fasta
結果です。
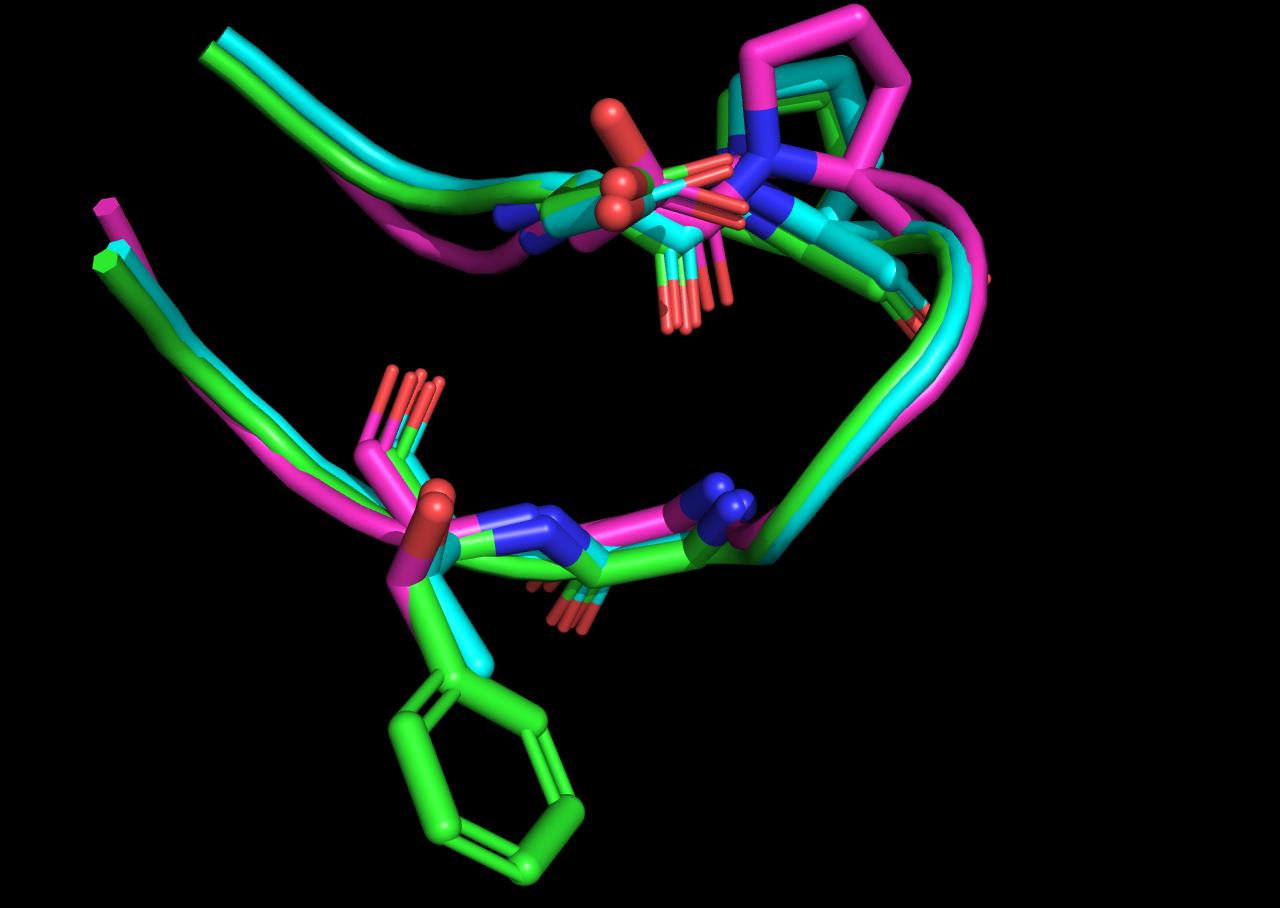
変異を入れてもboltz2の予測する構造は変化しなかったですね(cyan vs. green)。あとはPROのあたりの構造が結晶構造とは異なっていた。
機械学習の"発見"とか"新規"っていうのは既存の知識体系の中の人間の見落としを拾ったという意味であって、未知を開拓したっていう意味での発見ではないよなーっていうのが現在の私の立ち位置かなぁ
29062025 chemoinformatics
軽い気持ちで手元のMacbook Airでcrambin(46残基)の構造最適化をかけたら1時間かからずに計算が終了して「お、これは!」となったので、Facioで作られたインプットを見てみることにした。
!*** FMO 5.5 (Gamess) INPUT generated by Facio 27.1.1.64 ***
$CONTRL RUNTYP=OPTFMO NPRINT=-5 ISPHER=-1 MAXIT=50 $END
$SYSTEM MWORDS=58 $END
$GDDI NGROUP=1 $END
$SCF DIRSCF=.TRUE. NPUNCH=0 $END
$BASIS GBASIS=DFTB $END
! 基底関数はDFTBで固定っぽい
$DFTB
! DAMPXH = a flag to include the damping function for X-H atomic pair in DFTB3.
! DAMPEX = an exponent used in the damping function for X-H
! atomic pairs. The default value is 4.0 (taken from the 3OB parameter set).
! GAMESS includes 3OB-3-1, MATSCI03 (properly called matsci-0-3),
! and OB3W0PT3 (ob3 with omega = 0.3), which you may specify in PARAM.
SCC=.TRUE. DFTB3=.TRUE. DAMPXH=.TRUE. DAMPEX=4.00
PARAM=3OB-3-1
$END
!*** NumCPU : 1 MemPerNode : 512MB
$OPTFMO
! HSSUPD numeric updates of the inverse Hessian Default: HSSUPD.
METHOD=HSSUPD NSTEP=2000 OPTTOL=1E-4
$END
$PCM SOLVNT=WATER IEF=-10 ICOMP=0 ICAV=1 IDISP=1 MODPAR=65 IFMO=-1 $END
$PCMCAV RADII=SUAHF $END
$TESCAV NTSALL=60 $END
$FMOPRP
! MODPAR = parallel options
MODPAR=8205
NAODIR=210
NGRFMO(1)=1, 1, 0, 0, 0, 0, 0, 0, 0, 0
NPRINT=9
$END
$FMO
! MODGRD=2+8+32
! MODMUL Use the multipole expansion to compute electrostatic,
! interactions exactly bit additive.
! MAXBND = the maximum number of detached bonds. (default: NFG*2+1)
SCFTYP(1)=RHF
MODGRD=42 MODESP=0
MODMUL=0
MAXCAO=5
MAXBND=46
NLAYER=1
NFRAG=43
ICHARG(1)=
07052025 chemoinformatics life
Two things made me really happy. First, these articles(1,2) showed me how to preprocess PDB files for FMO calculations in a straightforward way. Second, thanks to faster CPUs these days, I found that FMO calculations on my MacBookAir with about 300 residues can now finish in just one day. That’s pretty amazing—20 years ago, it would have taken nearly a week using around 20 cluster machines!
An example is here

02052025 chemoinformatics life
After repeatedly rewriting input generators for FMO calculations each time I changed jobs, I decided to develop and maintain them as open-source software — and that’s how FMOkit was born.
To successfully carry out FMO calculations, we must overcome three major challenges:
In the following sections, we’ll examine each of these in detail.
Precompiled binaries are available for Windows and Intel-based Mac. On Linux, compilation is required, but the process is relatively straightforward. For Macs with Apple Silicon (M1–M3 chips), modifications to the source code are necessary. Please refer to the following Japanese article for detailed instructions.
FMO (Fragment Molecular Orbital) calculations are a type of quantum chemical simulation, and thus require that hydrogen atoms and fragment charges be explicitly included in the molecular system. However, standard structural formats such as PDB files typically lack this information, so it must be added during preprocessing.
While commercial molecular modeling tools like Maestro or MOE are often used in pharmaceutical companies and academic labs, I opted for an open-source solution. After evaluating various options, I chose OpenMM, a molecular dynamics toolkit, as a backend for preprocessing.
You can use the script mmcifutil.py located in the utils directory to add hydrogen atoms and assign partial charges to all atoms:
python mmcifutil.py input_file output_file
For details about how the code works, please refer to this article.
Aside from securing computational resources, generating input files for FMO calculations is arguably the most complex and unintuitive part of the entire workflow. Creating these inputs manually is not only extremely difficult, but also arguably inhumane in terms of workload and error-proneness.
Motivated by the desire to automate FMO input generation from structural data in the mmCIF format (with planned support for mol2 in the future), I began developing FMOkit.
If you prefer a graphical interface for preparing input files, I recommend using Facio, a GUI tool specifically designed for FMO workflows.
The code is shown below, using chignolin, one of the smallest known proteins, as an example.
>>> from FMOkit import System >>> s = System(nodes=1, cores=8, memory=12000, basissets="6-31G") >>> s.read_file("tests/5awl-addH.cif") >>> s.prepare_fragments() >>> with open("5awl-fmokit.inp", "w") as f: ... f.write(s.print_fmoinput())
Although command-line interface (CLI) support is planned in the future, the code was executed from the Python console for verification purposes in this example.
The FMO calculation (RHF/MP2, 6-31G basis sets) for this 10-residue protein completed in approximately 40 minutes on my MacBook Air (Apple M3, 2024, with 16 GB of memory).
% time ~/gamess/rungms 5awl-fmokit.inp >& t5awl-fmokit.out ~/gamess/rungms 5awl-fmokit.inp >&5awl-fmokit.out 2351.21s user 142.53s system 99% cpu 41:44.37 total
29042025 chemoinformatics
fmoutilをコンパイルする必要があったのだけど
gfortran fmoutil.f -o fmoutil
だとエラーが出るので一つオプションを追加する必要があった。
gfortran -std=legacy fmoutil.f -o fmoutil
それにしてもfortranは古のコードをよくコンパイルできるなぁと感心するのと、またコレの日々が繰り返されるのかとちょっと憂鬱ではある。
27042025 chemoinformatics work life
To create an input file for FMO calculations, we need both the structural information of the protein with hydrogen atoms included and the charge information for each fragment. However, since PDB files do not contain hydrogen atoms by default, we must add them using an external tool. Additionally, because the PDB format does not provide a field for storing partial charges, we repurposed the temperature factor field for this purpose.
Since the mmCIF format allows partial charge attributes to be added without unnecessary hacks, I wrote the code in openMM and gemmi.
from openmm.app import * from openmm import * from openmm.unit import * from sys import stdout import gemmi pdb = PDBFile("1AKI.pdb") forcefield = ForceField('amber14-all.xml', 'amber14/tip3pfb.xml') modeller = Modeller(pdb.topology, pdb.positions) residues=modeller.addHydrogens(forcefield) system = forcefield.createSystem( modeller.topology, nonbondedMethod=PME, nonbondedCutoff=1.0*nanometer, constraints=HBonds) partial_charges = [] for force in system.getForces(): if isinstance(force, openmm.NonbondedForce): for atom_index in range(force.getNumParticles()): charge, sigma, epsilon = force.getParticleParameters(atom_index) partial_charges.append(str(charge)[:-2]) with open('tmp.cif', 'w') as f: PDBxFile.writeFile(modeller.topology, modeller.positions, f) doc = gemmi.cif.read('tmp.cif') block = doc.sole_block() loop = block.find_loop('_atom_site.type_symbol').get_loop() loop.add_columns(['_atom_site.partial_charge'], value='?') pcs = block.find_values('_atom_site.partial_charge') for n, x in enumerate(pcs): pcs[n] = partial_charges[n] doc.write_file('output.cif')
I’m getting tired of writing FMO input generators every time I change jobs, so I’ve decided to upload them to GitHub as open-source software.
24122024 chemoinformatics
創薬 Advent Calendar 2024の24日目の記事です。久しぶりに書いてます。
以前にアドベントカレンダーに投稿してその勢いで入門書を書きました。 おかげさまで200超えの★と90超えのforkを得られたのでまずまず貢献したのかな?と思います。
その後、CBIのケモインフォマティクスハンズオンを開催することになり、そのマテリアルもオープンにしているので興味があれば一度動かしてみてください。
一般的なヴァーチャルスクリーニング(VS)を行うための入門的なハンズオン
化合物データの前処理(脱塩等)を行い、訓練データから活性予測モデルを作成。その後VS用のライブラリを構築し、Q活性予測モデルを利用しリガンドベースのヴァーチャルスクリーニング(LBVS)を行うという内容です。
こちらは中級以上向けで、完走率は3割切るくらいだったようですが、DRYの製薬企業研究者の2,3年目が到達するレベル感だと思います(私見)。こちらは化合物の生成モデルを使っているので色々応用が効く内容になっているはずです。ワークフローはコードで書いてもKNIME/PipelienPilotにまかせてもどっちでもいいような気がしますがバージョン管理するなら前者ですかね
来年やるとしたらALとかHITL関連のものをやってみたいですね。この論文にあるようにエキスパートを投入して環境に手をいれるっていうAZのアイデアも面白いですし、
Human-in-the-loop active learning for goal-oriented molecule generation https://t.co/tSxLzcF1dI #chemoinformatics #feedly
— kzfm (@fmkz___) December 23, 2024
または、Exscientiaのように生成モデルに工夫をしてRNN+RLじゃなくてビルディングブロックと反応をうまく学習させてより現実的な構造発生させるのにAL使うっていう感じの内容もよいかなと思ってます。
それではよいクリスマスを
14102024 chemoinformatics
CBI年会の初日のチュートリアルセッション(TS03 ケモインフォマティクスハンズオン)のリハをおこないました。
今回は去年のように最終調整を兼ねたリハ兼ビールでも飲んで楽しむというゆるいものではなく、課題の洗い出し兼あと二週間で潰すべきタスクのリストアップに近いような作業でした。なので結構疲れたけど、このタイミングでやっておいて本当に良かったと思いました(やらんかったら、相当グダグダなセッションになっているはず)。
わたしもmac担当として、REINVENT4きちんとインストールできるように試行錯誤したり、maizeがmacでも動くようにプルリク出したりと久々に働きました。
ハンズオンの流れとしては
という中級以上向けの攻めた内容になっていると思います。ちなみに、チュートリアルセッションは定員に達したそうで参加したい方はキャンセル待ちになるのではないでしょうか?
終了後にビールを飲みました(お約束)が、やはり来年以降はRDKitUGMJPの資金を調達しながら、ボランティアでやっているモデレーターにとってもwin-winになるような仕組みを考えたいなーと思いました。
14102024 chemoinformatics
As it could not be installed with the original requirements, I modified it and uploaded it to the Mishima-syk repository.
$ git clone https://github.com/Mishima-syk/REINVENT4.git $ cd REINVENT4 $ conda create --name reinvent4 python=3.11 $ conda activate reinvent4 $ pip install -r requirements-macOS.lock $ pip install --no-deps .
Now, you can set mps (Metal Performance Shaders) in the device parameter
# mps_sampling.toml # REINVENT4 TOML input example for sampling run_type = "sampling" device = "mps" # M3 GPU json_out_config = "_sampling.json" # write this TOML to JSON [parameters] ## Reinvent: de novo sampling model_file = "priors/reinvent.prior" output_file = 'sampling.csv' # sampled SMILES and NLL in CSV format num_smiles = 157 # number of SMILES to be sampled, 1 per input SMILES unique_molecules = true # if true remove all duplicatesd canonicalize smiles randomize_smiles = true # if true shuffle atoms in SMILES randomly
Running the reinvent4 program
$ reinvent -l sampling.log mps_sampling.toml