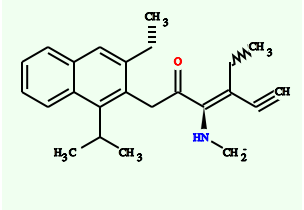
12082010 chemoinformatics Python
openbabelにはMaximum Common Substructure(MCS)を求めるメソッドがないのでGastonを使った。
似たようなのは昔書いたけど扱いにくいのでクラスにしといた。ついでにMCSSTanimotoも求めるようにした。
MCSSTanimoto(A,B) = MCS /(A + B - MCS) #全てヘテロ原子数
という、MCS版のタニモト距離。例えばOc1ccccc1 とCc1ccccc1のMCSTanimotoは
6 /(7 + 7 - 6) = 0.75
スキャフォールドが決まっている場合には割と手軽に使えんじゃないかなぁ。まぁヘテロの性質無視して同一性だけで判断しているのでハロゲンの違いは割と似ているとかそういう補正は入れたほうがいいのかもしれないが。
import openbabel as ob
import os,re
from tempfile import mkstemp
class Gaston:
def __init__(self,mols=[]):
self.mols = mols
def writefile(self,filename):
""" convert file to gaston format"""
f = open(filename, 'w')
for (molnum, mol) in enumerate(self.mols):
f.write("t # %d\n" % molnum)
for i,atom in enumerate(ob.OBMolAtomIter(mol)):
f.write("v %d %d\n" % (i,atom.GetAtomicNum()))
for i,bond in enumerate(ob.OBMolBondIter(mol)):
f.write("e %d %d %d\n" % (bond.GetBeginAtomIdx()-1,bond.GetEndAtomIdx()-1,bond.GetBondOrder()))
return filename
def readfile(self,file):
txt = open(file).read()
p = re.compile('#.+?(?=(#|$))',re.S)
mols = p.finditer(txt)
result = [self._gas2ob(gas.group()) for gas in mols]
return result
def _gas2ob(self,gf):
mol = ob.OBMol()
for l in gf.split('\n'):
if len(l) > 0 and l[0] == 'v':
a = mol.NewAtom()
atomic_num = int(l.split(' ')[2])
a.SetAtomicNum(atomic_num)
elif len(l) > 0 and l[0] == 'e':
begin_atom_idx = int(l.split(' ')[1]) + 1
end_atom_idx = int(l.split(' ')[2]) + 1
bond_order = int(l.split(' ')[3])
b = mol.AddBond(begin_atom_idx, end_atom_idx, bond_order)
elif len(l) > 0 and l[0] == '#':
title = l.split(' ')[1]
mol.SetTitle(title)
return mol
def search(self,freq=None):
if freq == None: freq = len(self.mols)
(m,gasfile) = mkstemp()
(n,output) = mkstemp()
self.writefile(gasfile)
os.system("/opt/local/bin/gaston %d %s %s > /dev/null 2>&1" % (freq,gasfile,output))
mols = self.readfile(output)
os.unlink(gasfile)
os.unlink(output)
return mols
def mcs(self):
substructures = self.search()
mcs = substructures[0]
for substructure in substructures[1:]:
if substructure.NumAtoms() > mcs.NumAtoms():
mcs = substructure
elif substructure.NumAtoms() == mcs.NumAtoms():
if substructure.NumBonds() > mcs.NumBonds():
mcs = substructure
return mcs
class MCSSTanimoto:
def __init__(self,mol1,mol2):
self.mol1 = mol1
self.mol2 = mol2
def score(self):
mcs = Gaston(mols=[self.mol1,self.mol2]).mcs()
return float(mcs.NumAtoms()) / (mol1.NumAtoms() + mol2.NumAtoms() - mcs.NumAtoms())
if __name__ == '__main__':
import sys
obc = ob.OBConversion()
obc.SetInAndOutFormats("smi", "smi")
mol1 = ob.OBMol()
obc.ReadString(mol1, "CCC1=CC=CS1")
mol2 = ob.OBMol()
obc.ReadString(mol2, "OCC1=CC=CS1")
mol3 = ob.OBMol()
obc.ReadString(mol3, "CCCC1=CC=CS1")
gaston = Gaston(mols=[mol1,mol2,mol3])
print ":::Frequent Structures:::"
for m in gaston.search():
sys.stdout.write(obc.WriteString(m))
print ":::MCS:::"
sys.stdout.write(obc.WriteString(gaston.mcs()))
print ":::MCSSTanimoto:::"
score = MCSSTanimoto(mol1,mol2).score()
print score
実行結果
:::Frequent Structures:::
CC 3
CC=C 3
C(=C)C=C 3
C(=C)C=CS 3
CC(=C)S 3
CC=CC 3
CC(=CC)S 3
CC=CC=C 3
CC(=CC=C)S 3
Cc1cccs1 3
CC=CC=CS 3
CC=CS 3
CC=CSC 3
c1ccsc1 3
CC=C(SC)C 3
CC=CSCC 3
CCS 3
CCSC 3
CC(=C)SC 3
CCSC=C 3
CC(=C)SC=C 3
C=C 3
C=CS 3
C=CSC 3
C=CSC=C 3
CS 3
CSC 3
:::MCS:::
Cc1cccs1 3
:::MCSSTanimoto:::
0.75
あと実際にモジュール化する場合にはコマンドがどこにあるかわからないし、もしかしたたらインストールされてないかもしれないけど、そういう場合にどういう感じで処理すればいいんだろうか?whichで調べるとかすんのかな?
 Rによるテキストマイニング入門
Rによるテキストマイニング入門 Quantum Chemical Reactivity Descriptors in Computational Drug Design
Quantum Chemical Reactivity Descriptors in Computational Drug Design Design and Use of Relational Databases in Chemistry
Design and Use of Relational Databases in Chemistry Optimizing the "Drug-Like" Properties of Leads in Drug Discovery (Biotechnology: Pharmaceutical Aspects)
Optimizing the "Drug-Like" Properties of Leads in Drug Discovery (Biotechnology: Pharmaceutical Aspects)