
14052010 chemoinformatics R
最近ダラけたというか、やる気が下がっていたところ、stochastic proximity embeddingとつぶやかれていたので、おー面白そうと調べたらpdfがあったので読んでみた。
多次元尺度法同様に、距離情報から座標を構成する手法らしい。ペーパーだと次元縮約の方法として紹介されているけど、一回距離情報を求めてそれを任意の次元に置き直すのでまぁ似たようなものかなと。
ただ、SPEのほうはデータセットが大きくなっても計算量が爆発しないので大きいデータに使えるそうだ。
- マップする次元を決めたら初期値としてランダムに座標を与える
- ランダムに二点を選ぶ(xi, xj)
- 距離が一致するように遠ければお互いを近づけ、短ければお互いを離す
- 何回か二点を選んで更新処理を行ったらラムダの値を小さくして、上を又繰り返す
ほーこれで上手くいくんかいなと思ったのだが、プラクティカルにはよさそうかも。でも、ベストな構造におちないのと、ラムダの刻み幅は小さくしないといけないっぽいな。 ちなみに、CRANにもspeというパッケージがあったので、今回これを動かしてみた。
library(spe)
data(phone)
embed <- spe(phone, edim=2, evalstress=TRUE)
plot(embed$x)
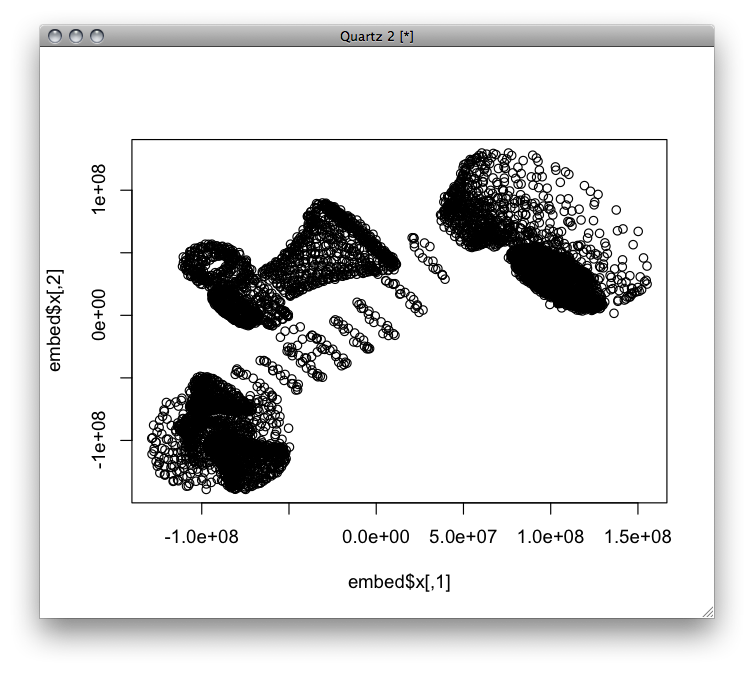
距離行列から計算できると便利だけどこのパッケージは座標をインプットにしなきゃいけないのとユークリッド距離固定な感じですね。実用で使う場合は自分で実装した方が思い通りに動かせるかな。あとアルゴリズムが単純なので並行処理もできるような気がするけど。そのうちScalaで書いてみたい感じ。

 Scalaスケーラブルプログラミング[コンセプト&コーディング] (Programming in Scala)
Scalaスケーラブルプログラミング[コンセプト&コーディング] (Programming in Scala) CouchDB: The Definitive Guide
CouchDB: The Definitive Guide Beginning CouchDB
Beginning CouchDB Couchdb in Action
Couchdb in Action

 Preclinical Development Handbook: ADME and Biopharmaceutical Properties (Pharmaceutical Development Series)
Preclinical Development Handbook: ADME and Biopharmaceutical Properties (Pharmaceutical Development Series) Software Design ( ソフトウェアデザイン ) 2010年 02月号 [雑誌]
Software Design ( ソフトウェアデザイン ) 2010年 02月号 [雑誌] Pharmacokinetic-Pharmacodynamic Modeling and Simulation
Pharmacokinetic-Pharmacodynamic Modeling and Simulation Pharmacometrics: The Science of Quantitative Pharmacology
Pharmacometrics: The Science of Quantitative Pharmacology Evaluation of Drug Candidates for Preclinical Development: Pharmacokinetics, Metabolism, Pharmaceutics, and Toxicology (Wiley Series in Drug Discovery and Development)
Evaluation of Drug Candidates for Preclinical Development: Pharmacokinetics, Metabolism, Pharmaceutics, and Toxicology (Wiley Series in Drug Discovery and Development)