MOROKUMA ANALYSISのサンプル(EXAM28)を。
水とアンモニアのダイマー計算
$contrl scftyp=rhf runtyp=morokuma coord=zmt $end
$basis gbasis=n31 ngauss=4 $end
$guess guess=huckel $end
$morokm iatm(1)=3 $end
$data
water-ammonia dimer
Cs
H
O 1 rOH
H 2 rOH 1 aHOH
N 2 R 1 aHOH 3 0.0
H 4 rNH 3 aHNaxis 1 180.0
H 4 rNH 3 aHNaxis 5 +120.0
H 4 rNH 3 aHNaxis 5 -120.0
rOH=0.956
aHOH=105.2
rNH=1.0124
aHNaxis=112.1451 ! makes HNH=106.67
R=2.93
$end
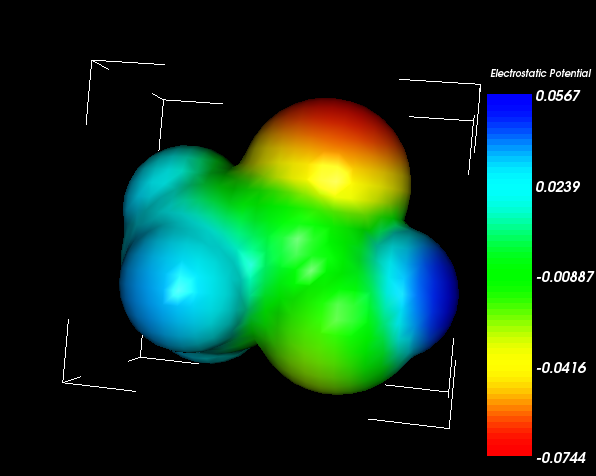
結果
HARTREE KCAL/MOLE
ELECTROSTATIC ENERGY ES= -0.022336 -14.02
EXCHANGE REPULSION ENERGY EX= 0.014308 8.98
POLARIZATION ENERGY PL= -0.001786 -1.12
CHARGE TRANSFER ENERGY CT= -0.003780 -2.37
HIGH ORDER COUPLING ENERGY MIX= -0.000687 -0.43
TOTAL INTERACTION ENERGY, DELTA-E= -0.014282 -8.96
DECOMPOSITION OF CT
CHARGE TRANSFER ENERGY, MON= 1 CT= -0.000494 -0.31
CHARGE TRANSFER ENERGY, MON= 2 CT= -0.003286 -2.06
DECOMPOSITION OF PL
EPL, MON= 1 PL= -0.001068 -0.67
EPL, MON= 2 PL= -0.000584 -0.37
HIGH ORDER COUPLING FOR PL, PMIX= -0.000133 -0.08
複合体結晶構造から重要な残基とのインタラクションを見積もるのに、リガンドと特定の 残基のシンプルなモデルを切り出してきて、きちんと計算すればよかろうと。
 すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)
すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)