------------------------------------
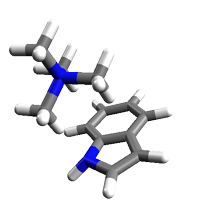
RESULTS OF KITAURA-MOROKUMA ANALYSIS
------------------------------------
HARTREE KCAL/MOLE
ELECTROSTATIC ENERGY ES= -0.011414 -7.16
EXCHANGE REPULSION ENERGY EX= 0.008788 5.51
POLARIZATION ENERGY PL= -0.001679 -1.05
CHARGE TRANSFER ENERGY CT= -0.007053 -4.43
HIGH ORDER COUPLING ENERGY MIX= 0.000166 0.10
TOTAL INTERACTION ENERGY, DELTA-E= -0.011193 -7.02
水素結合と同じくらいの強さかな。
以下、STO-3Gで構造最適化した座標を使ってつくったインプット
$BASIS GBASIS=STO NGAUSS=3 NDFUNC=1 $END
$CONTRL SCFTYP=RHF RUNTYP=MOROKUMA MAXIT=200 ICHARG=1 MULT=1 $END
$MOROKM IATM(1)=17 ICHM(1)=1 $END
$DATA
W188_BET.out
C1
N 7.0 26.56861 22.88472 72.47206
C 6.0 25.05591 23.05763 72.38527
C 6.0 27.18276 23.04159 71.08205
C 6.0 27.14794 23.94295 73.40648
C 6.0 26.89124 21.49601 73.01604
H 1.0 24.65761 22.29324 71.71973
H 1.0 24.84300 24.05067 71.99229
H 1.0 26.95089 24.04166 70.71972
H 1.0 26.73892 22.29197 70.42917
H 1.0 28.26387 22.89551 71.15590
H 1.0 26.89847 24.92550 73.00913
H 1.0 28.22884 23.81270 73.44995
H 1.0 26.70726 23.81001 74.39332
H 1.0 26.46855 20.75492 72.33939
H 1.0 26.44825 21.40230 74.00640
H 1.0 27.97398 21.38820 73.06968
H 1.0 24.63622 22.94695 73.38408
C 6.0 30.61324 20.49644 72.40614
C 6.0 30.47183 21.03406 71.19420
C 6.0 30.79531 21.59051 73.36029
C 6.0 30.75216 22.77934 72.62888
C 6.0 31.00908 21.63948 74.74382
N 7.0 30.43745 22.45766 71.27884
C 6.0 30.92671 24.02761 73.23103
C 6.0 31.18435 22.86580 75.34238
C 6.0 31.14962 24.05129 74.58925
H 1.0 30.33993 20.55500 70.23268
H 1.0 31.04481 20.72825 75.32842
H 1.0 30.86231 23.00634 70.52231
H 1.0 30.90815 24.93911 72.64846
H 1.0 31.36065 22.92555 76.40870
H 1.0 31.30378 24.99933 75.08996
H 1.0 30.61446 19.44515 72.64676
$END


 エキスパートPythonプログラミング
エキスパートPythonプログラミング


 みんなのPython 改訂版
みんなのPython 改訂版 すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)
すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)