
12012010 chemoinformatics perl Python
bioinformaticsの学習道路はそこそこ充実している。
でも、chemoinformaticsだってオープンソースで色々できるし、独学でも結構いけるよ!なんて思うんだけど情報が足りないなぁと思った。
というわけで、ちょっと小道でも。
特にperl,pythonでちょっとした化学情報をいじるコードをクックブック形式で書いていけたらなぁなんて思ってる。
週に一回くらいは更新するつもりで。
12012010 chemoinformatics perl Python
bioinformaticsの学習道路はそこそこ充実している。
でも、chemoinformaticsだってオープンソースで色々できるし、独学でも結構いけるよ!なんて思うんだけど情報が足りないなぁと思った。
というわけで、ちょっと小道でも。
特にperl,pythonでちょっとした化学情報をいじるコードをクックブック形式で書いていけたらなぁなんて思ってる。
週に一回くらいは更新するつもりで。
12012010 chemoinformatics Python
MolBlasterは細切れにして、頻度を返さないといけないんだが、Separateメソッドがバグッてるらしいので、canonical smilesで出してpythonのほうで.でsplitして頻度をみてみた
import openbabel as ob
from random import sample
def randomsplit(mol,cutnum=5):
cutlist = sample(range(mol.NumBonds()),cutnum)
delbonds = []
for i,bond in enumerate(ob.OBMolBondIter(mol)):
if i in cutlist: delbonds.append(bond)
for b in delbonds: mol.DeleteBond(b)
return mol
def molblast(smi,iter=100,cutnum=5):
obc = ob.OBConversion()
obc.SetInAndOutFormats('smi','smi')
obc.AddOption('c',ob.OBConversion.OUTOPTIONS)
freq = {}
for i in range(iter):
mol = ob.OBMol()
obc.ReadString(mol,smi)
for fragment in obc.WriteString(randomsplit(mol,cutnum=cutnum))[:-2].split('.'):
freq[fragment] = freq.get(fragment,0) + 1
return freq
if __name__ == "__main__":
smiles = 'CCC(C)C(=O)OC1CC(C=C2C1C(C(C=C2)C)CCC(CC(CC(=O)O)O)O)O'
print molblast(smiles,iter=1000,cutnum=20)
こんな感じ。
{'CCCCC=CC': 2, 'O=COCCC': 1, 'CCC=C(C)C(C)CC': 1, 'CCC(=O)O': 26, 'CCCCC(C)O':
2, 'CCO': 459, 'CC=CCC(C)C': 2, 'CCC=CC(C)O': 2, 'CCCCC(C)C': 4, 'CCC': 705,
'CCC=CC=CCC': 1, 'OCCCCC': 10, 'OCCC(C)OC': 1, 'C=CC=CCC': 2, 'CCCC(=C)C': 3,
...
細切れにしないと頻度が稼げないし、細切れにするとファーマコフォア的な構造特徴が失われてしまう感じだ。というわけで、やってみて分かったけど、多分この方法だと実用的じゃないなぁ。Fragment Dependency Graphとかは面白そうなんだけどなぁ。
12012010 chemoinformatics Python openbabel
Molblasterっていうのは化学構造をランダムに切断していくっていう試行を何度か繰り返す方法で、その後フラグメントの頻度をシャノンエントロピーで評価したりするらしい。
シャノンエントロピーのほうに興味があったのとちょっと簡単に実装できそうだったのでpython+openbabelでやってみた。
まぁでも、構造活性相関にもっていくんだったら普通はベイズの方にいくよなぁ。多分著者の論文あると思うんだけど調べてない。
12012010 chemoinformatics Python
Topological Fragment Indexというものが気になったので。
論文はこれでToFIを尤度として捉えているらしい。
式は
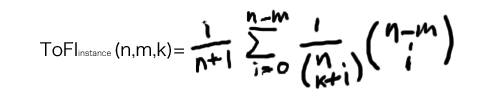
ここで、nは全ボンド数。kはフラグメントについてる余計なボンド。lをフラグメントのボンド数としてm=k+lとなっている。
def combination(n,m):
if m == 0:
return 1.0
else:
return reduce(lambda x,y:x*y,range(n-m+1,n+1)) / float(reduce(lambda x,y:x*y,range(1,m+1)))
def tofi(n,m,k):
sum = 0.0
for i in range(n-m+1):
sum += combination(n-m,i) / combination(n,k+i)
return sum/(n+1)
で、整数にするために1*10^6という定数をかけるらしいのだが、なにがしたいのかよくわからん。
lが一定でkが増えるとToFIが減る
>>> tofi.tofi(23,17,5) * 1000000
8.9779501544207427
>>> tofi.tofi(23,18,6) * 1000000
2.8351421540276029
>>> tofi.tofi(23,19,7) * 1000000
0.99229975390966119
kが一定でlが減るとToFIが増える
>>> tofi.tofi(23,17,5) * 1000000
8.9779501544207427
>>> tofi.tofi(23,16,5) * 1000000
13.466925231631114
>>> tofi.tofi(23,15,5) * 1000000
20.812520812520813
後者はなんとなく理解できるけど、切断面が大きくなるほどToFIが減るってのがわからん。
計算間違ってるのかなぁ。
12012010 chemoinformatics bioinformatics Python
NMFを追っかけてたらMetagenes and molecular pattern discovery using matrix factorizationという論文を見つけたので、週末はこの論文を読みながら色々やってみた。NMFの便利なところは元の特徴(この論文の場合は遺伝子発現量)からより少ない任意の特徴量(論文中ではmetagene)に変換できるところであり、さらにそのままクラスターの分割に利用できる。
たとえば2つのmetageneで表現した場合、より発現量の大きいmetageneで分割すれば2つのクラスに分けられる。(QSARだったらdescriptorからmeta discriptorが導かれてそれに基づいてクラス分類ができるでしょう)
続いて、重要なのがクラスの安定性である。要するに最適なクラスタの数はいくつなのかということである。これに対して、この論文ではConsensus Clusteringというリサンプリングと隣接行列(connectivity matrix)を利用する方法をモディファイした方法を使っている。
ここで隣接行列はi番目のサンプルとj番目のサンプルが同じクラスなら1、それ以外なら0である。この行列のn回の平均値をコンセンサスマトリックスとする。コンセンサスマトリックスの値は0-1の間をとり、サンプルi,jが常に同じクラスになる場合は1、常に異なるクラスの場合は0である。フラフラするばあいはその間の値をとる。元の論文のconsensus clusteringアルゴリズムはデータの80%をランダムサンプリングして評価するのに対し、NMFの場合、初期値の行列はランダムな数字にしているので適当にループ(n)をまわすだけでよい。
NMFの実装は集合知プログラミングのものを用い、コンセンサスマトリックスを評価するコードをnumpyを使ってかいた。
import nmf
from numpy import *
def consensus(a,kstart,kend,nloop):
""" calculate consensus matrix
"""
(n,m) = a.shape
consensus = zeros((kend+1,m,m))
conn = zeros((m,m))
#i = 0
for j in range(kstart,kend+1):
connac = zeros((m,m))
for l in range(nloop):
#i += 1
(w,h) = nmf.factorize(a,pc=j)
conn = nmfconnectivity(h)
connac = connac + conn
consensus[j] = connac / float(nloop)
return consensus
def nmfconnectivity(h):
""" calculate connective matrix
"""
(k,m) = h.shape
ar = []
for i in range(m):
max_i = 0
max_v = 0
for index,v in enumerate(h[:,i]):
if v > max_v :
max_v = v
max_i = index
ar.append(max_i)
mat1 = tile(matrix(ar),(m,1))
mat2 = tile(matrix(ar).T,(1,m))
return array(mat1 == mat2, dtype=int)
これをcc.pyという名前で保存しテスト用のセットを適当に用意して実行。
from numpy import *
from pylab import *
import cc
kstart = 2
kend = 4
testarray = array([[0,0,1,1,1,0,0,0,0],
[0,0,1,1,0,0,0,0,0],
[1,1,0,0,1,1,0,0,1],
[1,1,0,0,0,0,0,1,0],
[1,1,0,1,0,0,0,0,0],
[0,0,0,0,0,1,1,1,1],
[1,0,0,0,0,1,1,1,0],
[0,0,0,0,0,0,1,1,1],
[0,0,0,1,0,1,1,1,1]
])
cons = cc.consensus(testarray.T,kstart,kend+1,20)
for i in range(kstart,kend+1):
pcolormesh(cons[i])
if i == kstart:
colorbar()
savefig('ccr' + str(i) + '.png')
見ればすぐ分かるが3つにクラスタリングできそうなマトリックス。
結果
2クラスタで分けた場合。
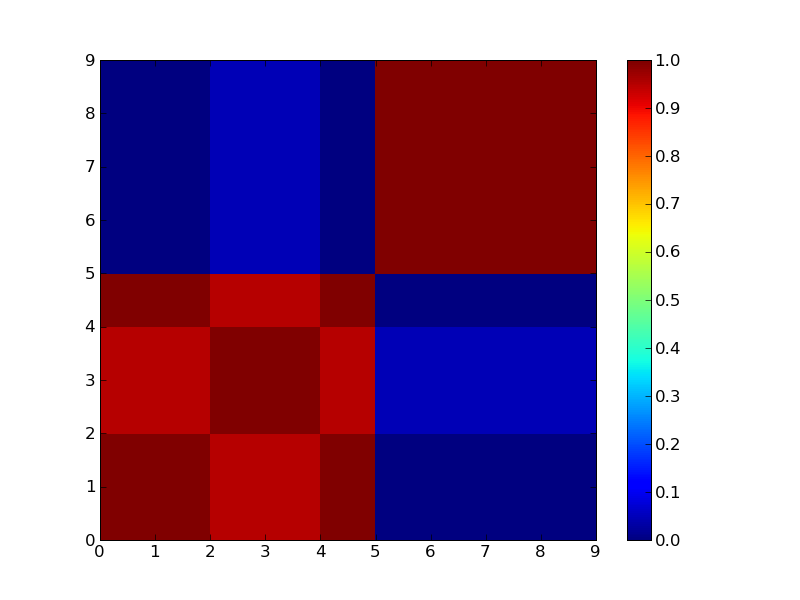
3クラスタで分けた場合
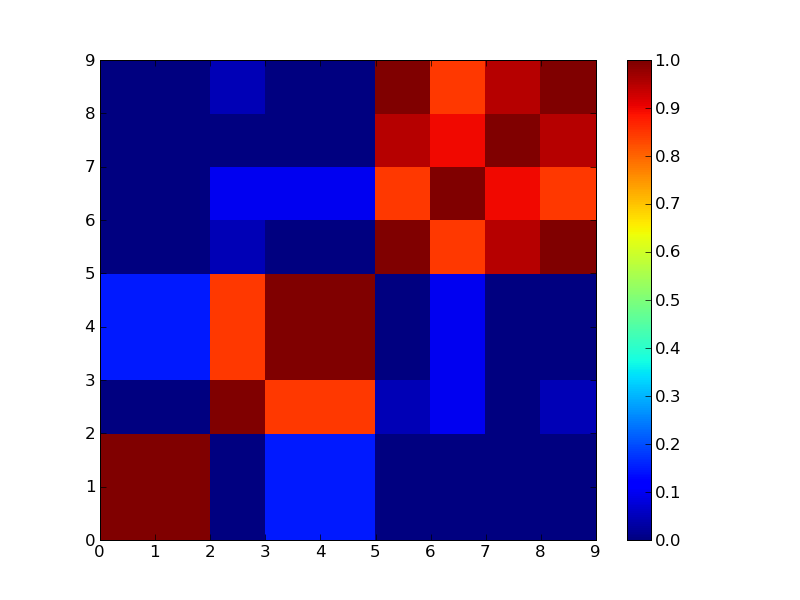
4クラスタで分けた場合
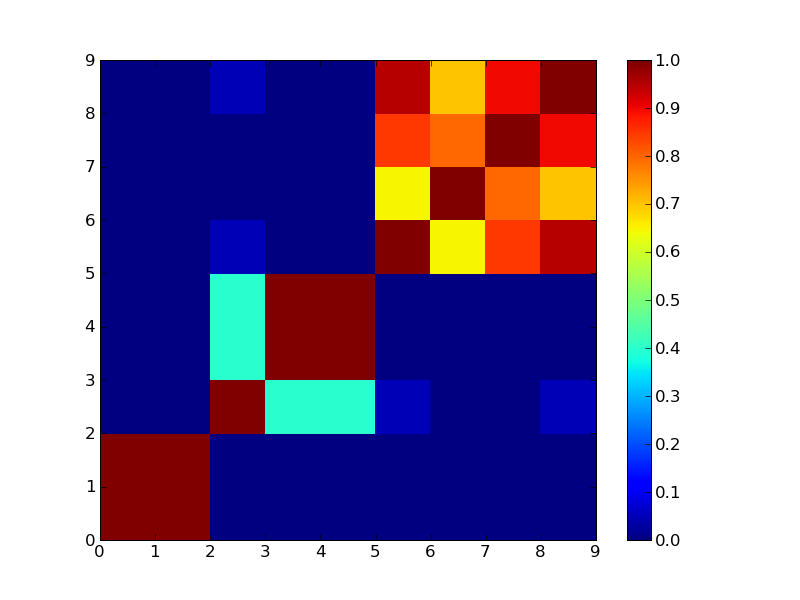
もうちょっと実際のデータでやってみないとあれだなぁ。それとConsensus ClusteringのCDFプロットみたいなのが欲しいところ。
参考書籍
参考論文
12012010 Python matplotlib
ちょっと描きたいものがあったので調べたらpcolormeshを使えばいいだけだった。
from numpy import *
from pylab import *
a = rand(100,100)
pcolormesh(a)
colorbar()
savefig('colour.png')
12012010 bioinformatics Python
12012010 Python
X-meansの論文を眺めていたら、最初から2-meansやってBICが大きくなる限り再帰的に2-meansやればいいんじゃないの?と思ったので、そういう風なものを書いてみたという。
K-meansのコードは「集合知プログラミング」のものを参考にした。データも集合知プログラミングのブログのデータを使ってます
コードは分散のとことか、結局クラスのデータが一つ二つになっちゃった場合とかのエラーに対応すんのがめんどくなって適当にいじってるのでかなりいい加減というかいい加減すぎなのでダメすぎなので、そういうかんじで。BICもきちんと理解しないといけないのだけど、とりあえずやっつけ的になってしまった。
import random
from math import sqrt,pi,log
def pearson(v1,v2):
sum1=sum(v1)
sum2=sum(v2)
sum1Sq=sum([pow(v,2) for v in v1])
sum2Sq=sum([pow(v,2) for v in v2])
pSum=sum([v1[i]*v2[i] for i in range(len(v1))])
num=pSum-(sum1*sum2/len(v1))
den=sqrt((sum1Sq-pow(sum1,2)/len(v1))*(sum2Sq-pow(sum2,2)/len(v1)))
if den==0: return 0
return 1.0-num/den
def euclid(v1,v2) :
return sqrt(sum([pow(v1[i]-v2[i],2) for i in range(len(v1))]))
def kcluster(rows,cluster,distance=euclid,k=2):
ranges=[(min([rows[c][i] for c in cluster]),max([rows[c][i] for c in cluster]))
for i in range(len(rows[0]))]
clusters=[[random.random()*(ranges[i][1]-ranges[i][0])+ranges[i][0]
for i in range(len(rows[0]))] for j in range(k)]
lastmatches = None
for t in range(100):
bestmatches=[[] for i in range(k)]
for j in cluster:
row = rows[j]
bestmatch = 0
for i in range(k):
d = distance(clusters[i],row)
if d < distance(clusters[bestmatch],row): bestmatch=i
bestmatches[bestmatch].append(j)
if bestmatches == lastmatches: break
lastmatches = bestmatches
for i in range(k):
avgs=[0.0]*len(rows[0])
if len(bestmatches[i])>0:
for rowid in bestmatches[i]:
for m in range(len(rows[rowid])):
avgs[m]+=rows[rowid][m]
for j in range(len(avgs)):
avgs[j]/=len(bestmatches[i])
clusters[i]=avgs
return bestmatches,clusters
def readfile(filename):
lines=[line for line in file(filename)]
colnames=lines[0].strip().split('\t')[1:]
rownames=[]
data=[]
for line in lines[1:]:
p=line.strip().split('\t')
rownames.append(p[0])
data.append([float(x) for x in p[1:]])
return rownames,colnames,data
def likelihood(data,cluster,avg_cluster,k):
sample_number = len(cluster)
variance = sum([pow(data[c][i] - avg_cluster[i],2) for i in range(len(avg_cluster)) for c in cluster]) / float(sample_number)
if variance == 0: variance = 0.0000000000000001
lkh = -sample_number/2*log(2*pi) - sample_number*len(avg_cluster)/2*log(variance) - (sample_number - k)/2 + sample_number*log(sample_number)
return lkh
def _xcluster(data,cluster,bic,k):
bm,cl = kcluster(data,cluster)
if len(bm[0]) == 0 or len(bm[1]) == 0:
bm,cl = kcluster(data,cluster)
new_likelihood = likelihood(data,bm[0],cl[0],2) + likelihood(data,bm[1],cl[1],2)
new_bic = new_likelihood - (2*len(data[0])+1)/2 * log(len(cluster))
if bic < new_bic:
if len(bm[0]) > 1:
bicl = likelihood(data,bm[0],cl[0],1) - (len(data[0])+1)*log(len(bm[0]))/2
nl = _xcluster(data,bm[0],bicl,2)
else:
nl = bm[0]
if len(bm[1]) > 1:
bicr = likelihood(data,bm[1],cl[1],1) - (len(data[0])+1)*log(len(bm[1]))/2
nr = _xcluster(data,bm[1],bicr,2)
else:
nr = bm[1]
return [nl,nr]
else:
return cluster
def xcluster(filename,distance=euclid,k=2):
rownames,colnames,data = readfile(filename)
sample_number = len(data)
params = len(data[0])
avg = [sum([data[j][i] for j in range(sample_number)])/float(sample_number) for i in range(params)]
bic = likelihood(data,range(len(data)),avg,1) - (params+1)*log(sample_number)/2
bestclusters = _xcluster(data,range(sample_number),bic,2)
return bestclusters
コードはいろんな部分がダメすぎてあれなんだけど、とりあえず実行。
>>> from xmeans import *
>>> xcluster("blogdata.txt")
[[[[[[[0, 2, 3, 5, 7, 8, 11, 12, 13, 15, 16, 17, 20, 21, 22, 26, 28, 29,
30, 32, 34, 36, 38, 39, 40, 43, 47, 48, 50, 51, 52, 53, 54, 55, 56, 57, 58,
59, 60, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 74, 75, 76, 77, 78, 79, 80,
83, 84, 86, 87, 88, 89, 92, 95, 96, 97], [44]], [[91], [37]]], [1, 6, 9,
10, 14, 18, 19, 27, 31, 33, 35, 45, 61, 73, 82, 90, 93, 98]], [[85], [[23],
[[25], [[[49], [94]], [62]]]]]], [[4], [[81], [[42], [[41], [46]]]]]], [24]]
あとで、もうちょっと考える。
12012010 chemoinformatics Python
12012010 Python
pythonの組み込みウェブサーバーは便利で
import SimpleHTTPServer
SimpleHTTPServer.test()
という打鍵をpythonのインタラクティブシェルを起動してやっているのだけどPython - Swiss Army Knife Web Serverのコメントにあるような、
python -m SimpleHTTPServer
なんてできることを初めて知った。ということで、次の一行を.bashrcに書いておけば
alias pyhttpd='python -m SimpleHTTPServer'
いつでもpython組み込みのサーバーが起動して快適だ。例えば、画像突っ込んだディレクトリの内容なんかはさくっと見れていい感じ。
-mで呼び出すとモジュールの__main__が呼ばれるのね。モジュールの中身をみたら
if __name__ == '__main__':
test()
って書いてあった。なるほど。(デーモンじゃないけど分かりやすいのでpyhttpdってコマンドで使うことにした。)
 集合知プログラミング
集合知プログラミング