さて、bioinformaticsとかchemoinformaticsとか言われてるようなアレは、化学とか生物学に対して情報学的観点からアプローチしたりするわけです。こういう点から見ると、薬ってもんは蛋白質にうまいことはまる鍵みたいなもんで、蛋白質の穴にいい感じではまるような化合物を(コンピュータを駆使して)設計していくのが(コンピュテーショナルな)ドラッグデザイン(CADD)という分野だヨ。
で、蛋白質というものは複数のアミノ酸から構成され(100から数百)そしてアミノ酸は20種類程度存在し、それぞれ数十の原子から構成されているわけだ。要するに一対多の階層構造をとる。
protein -> amino-acid -> atom
みたいな。
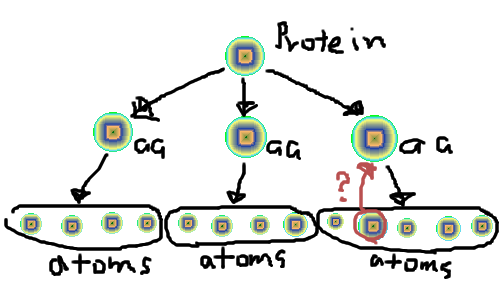
でそれぞれprotein aminoacid(aa) atomみたいなクラスを用意すれば
$atom1 = Atom->new({name => 'C1', type => 'C'});
...
$aa1 = AminoAcid->new({name => 'GLY', atoms => [$atom1, $atom2, ]})
...
$protein = Protein->new({name=> 'ProteinA', aminoacids => [$aa1,$aa2,$aa3...]});
みたいにそれぞれ配列に突っ込めば蛋白質を表現できて、あるatomオブジェクトを与えられた場合にそれがどのアミノ酸に属しているのか調べるのに
for my $aminoacid ($protein->aminoacids){
for my $atom ($aminoacid->atoms){
return $aminoacid if $qatom == $atom;
}
}
みたいにdepthfirstで探索していけばいいんだろうけど、ちょっと探索効率が悪いので、atomオブジェクトに $atom->{parent} = $parent_aminoacidみたいに親のアミノ酸オブジェクト返すような属性追加したんだけど、これだと構造が複雑になってなんか気持ち悪い。
他にうまいやり方ってあんのかなと思ったお盆の夏2007。

 R Commanderハンドブック―A Basic-Statistics GUI for R
R Commanderハンドブック―A Basic-Statistics GUI for R