
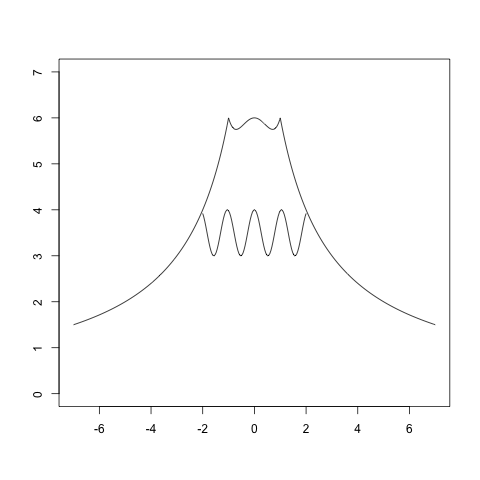
富士山をプロットする関数をみかけたので、Rで描いてみた。
fx <- function(x){x^4-x^2+6} sfx <- function(x){12/(abs(x)+1)} gx <- function(x){1/2*cos(6*x)+7/2} plot(gx,-2,2,xlim=c(-7,7),ylim=c(0,7),xlab="",ylab="") par(new=T) plot(sfx,-7,-1,xlim=c(-7,7),ylim=c(0,7),axes=F,xlab="",ylab="") par(new=T) plot(sfx,1,7,xlim=c(-7,7),ylim=c(0,7),axes=F,xlab="",ylab="") par(new=T) plot(fx,-1,1,xlim=c(-7,7),ylim=c(0,7),axes=F,xlab="",ylab="")
ggplotで関数だけ描く方法わからんなぁとつぶやいたら、教えてもらったのでggplot2でも描いた。
gx <- function(x){ x[-2>x | x>2] <-NA 1/2*cos(6*x)+7/2 } fx <- function(x){ x[x<(-1)] <- NA x[x>1] <- NA x^4-x^2+6 } fx <- function(x){ x[-1<x & x<1] <-NA 12/(abs(x)+1) } ggplot(data.frame(x=c(-7,7),y=0), aes(x,y))+stat_function(fun=gx) \ +stat_function(fun=fx)+stat_function(fun=sx)
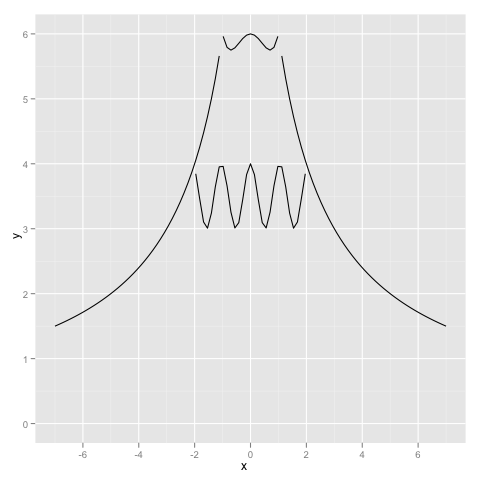
ggplot2だと線がうまくつながらなかった。
 Ggplot2: Elegant Graphics for Data Analysis (Use R!)
Ggplot2: Elegant Graphics for Data Analysis (Use R!)Hadley Wickham
Springer-Verlag New York Inc (C) / ¥ 5,259 ( 2009-08-30 )
 ネットワーク分析 (Rで学ぶデータサイエンス 8)
ネットワーク分析 (Rで学ぶデータサイエンス 8)
 Bioinformatics Programming Using Python
Bioinformatics Programming Using Python Statistical Bioinformatics: with R
Statistical Bioinformatics: with R Building Bioinformatics Solutions: With Perl, R and Mysql
Building Bioinformatics Solutions: With Perl, R and Mysql R Programming for Bioinformatics (Chapman & Hall/Crc Computer Science & Data Analysis)
R Programming for Bioinformatics (Chapman & Hall/Crc Computer Science & Data Analysis) Rの基礎とプログラミング技法
Rの基礎とプログラミング技法
 確率微分方程式―入門から応用まで
確率微分方程式―入門から応用まで