
17062009 q-chem
Molekelいい感じ。
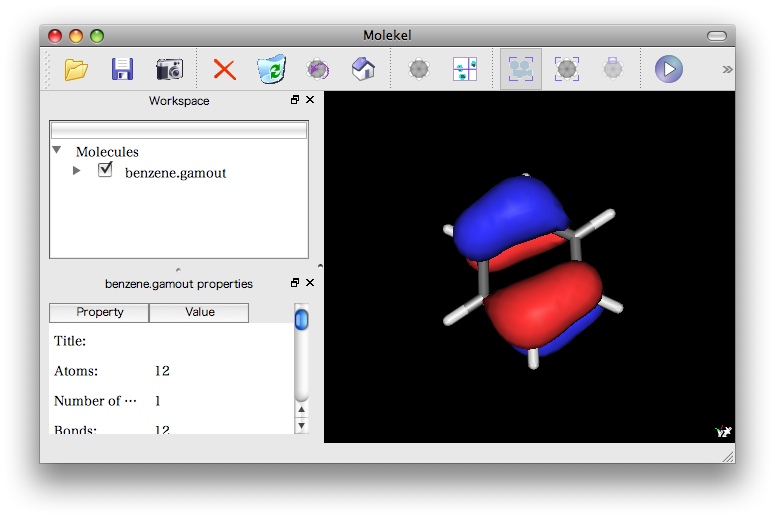
ありがちなエラー。
*** ERROR! THERE ARE NOT 5 OR 6 TRANS/ROT MODES NUM
そういう場合には
$statpt projct=.f. $end
と入れとく
こんな感じ?
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 DIFFSP=.TRUE. $END
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE DFTTYP=B3LYP NUMGRD=.T. $END
$SVP $END
$STATPT OPTTOL=0.0001 NSTEP=200 $END
$DATA
Title
C1
O 8.0 -4.21789 1.92792 0.00000
H 1.0 -3.25037 1.97435 0.00000
H 1.0 -4.49708 2.85544 0.00000
$END
多分だめだな。あとでやり直す
10062009 q-chem
最近、妙にまじめに取り組んでいる。
deltaGをもとめるために、気相と液相それぞれについてアニオンと、中性化合物のGを求めなければならん。というか化合物毎に4つも計算するのは面倒だ。
これには書いてないけど、最近の論文追っかけてみると、なかなか面白そうなことが書いてあったりする。量子化学系もオープンアクセスになればいいのに。
ふと、オープンソースのソフトウェアの多い学問からオープンアクセス化するのかなぁと思った。
あと、mimetex記法を導入しないと面倒で仕方ない。
GUESSがヒュッケルでよいのかなぁ?と。走らせてみたいだけなのでまぁよしとする。
$contrl scftyp=rhf runtyp=energy $end
$system mwords=4 $end
$basis gbasis=n31 ngauss=6 ndfunc=1 npfunc=1 $end
$guess guess=HUCKEL norb=16 $end
$scf nconv=8 $end
$svp nvlpl=3 rhoiso=0.001 dielst=78.304 nptleb=1202 $end
$data
CH3CONH2 cgz geometry RHF/6-31G(d,p)
C1
C 6.0 1.361261 -0.309588 -0.000262
C 6.0 -0.079357 0.152773 -0.005665
H 1.0 1.602076 -0.751515 0.962042
H 1.0 1.537200 -1.056768 -0.767127
H 1.0 2.002415 0.542830 -0.168045
O 8.0 -0.387955 1.310027 0.002284
N 7.0 -1.002151 -0.840834 -0.011928
H 1.0 -1.961646 -0.589397 0.038911
H 1.0 -0.752774 -1.798630 0.035006
$end
結果
SVPE SOLVATION METHOD ENERGIES
----------------------------------
SOLUTE INTERNAL ENERGY = -207.98571642
[SURFACE POLARIZATION ENERGY] = -0.02089990 ( -13.11489 KCAL/MOL )
[VOLUME POLARIZATION ENERGY] = 0.00033438 ( 0.20982 KCAL/MOL )
[VOL POL OUTER LAYER ENERGY] = 0.00000212 ( 0.00133 KCAL/MOL )
REACTION FIELD FREE ENERGY = -0.02056552 ( -12.90506 KCAL/MOL )
TOTAL FREE ENERGY IN SOLUTION = -208.00628195
rungmsをちょっと書き換える必要がある。 あとgmsにalias切った。
set TARGET=sockets
set SCR=/Users/kzfm/gamess/scr
set GMSPATH=/Users/kzfm/gamess
# location where the .dat and .irc files go
set USERSCR=/Users/kzfm/scr
#
set JOB=$1 # name of the input file xxx.inp, give only the xxx part
set VERNO=$2 # revision number of the executable created by 'lked' step
set NCPUS=$3 # number of compute processes to be run
# provide defaults if last two arguments are not given to this script
if (null$VERNO == null) set VERNO=Jan122009R1
if (null$NCPUS == null) set NCPUS=1
水の構造最適化用インプットファイル
$CONTRL RUNTYP=OPTIMIZE SCFTYP=RHF MULT=1 MAXIT=100 UNITS=ANGS
$END
$SYSTEM TIMLIM=10000 MWORDS=30 MEMDDI=10 $END
$BASIS GBASIS=N31 NGAUSS=6 NPFUNC=1 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$DATA
H2O RHF/6-31G(D P) OPTIMIZE
CNV 2
O 8.0 0.00000 0.00000 0.00000
H 1.0 0.87 0.00 -0.50
実行
gms h2o.inp > h2o.out
13042009 chemoinformatics q-chem
ちょっと来月の頭に講演をしなきゃいけなくなったので、色々と考えていたら 、僕のシステムバイオロジー的な世界観というものは、古代インドの宇宙観にわりと近いのではないかという結論に達した。

つまり、ゲノムは蛇であり、量子化学的世界とはこの世の全てを取り巻くトラパーの波であり、真理なのであった。
31082007 chemoinformatics q-chem
13082006 q-chem
Structure-Based Drug Designに従事していると、トラディッショナルな目視での解釈に限界と疑問を感じる時が遅かれ早かれやってくるはず。
そうなると、解いた蛋白-リガンド結晶構造をGAMESSとかABINIT-MPなんかでFMO計算し始めるわけだ(そのうちただ単に計算したいという理由でPDBのデータなんかに手出したり)これにより、電子的な相互作用をエネルギーの大小で考えることができるようになるため、複合体に対する理解が深まるのでイイ。
でも、やがてこれだけでは満足できなくなり、より深い解釈を求め始めるようになる。つまり、生化学、構造生物学から、有機化学、量子化学へと足を踏み入れはじめるようになるわけだ。
ただし、このような場合に、勉強するのにちょうどいい本や資料がないなぁと困っていたところ、GAMESSについて書かれている本が出版されたというエントリをみっけたので、脊髄反射的にamazonで購入してしまった。
s2k's eye 2nd Ed.: 「すぐできる量子化学計算…」を買いました。
Q&A形式で、どこからでも読めるというのも好印象です。そして、何より、今までの成書ではあまり無かったGAMESSについての解説が多く入れられています(GaussianとGAMESSの解説書です)。応用計算については多くがGaussianについてですが、これはGAMESSというプログラムの性質上しょうがないところでしょう。
内容は、FAQ集という感じで、200pぐらいある割には半日かからずに読み終えた。もちろんGAMESSに関する記述も結構あるので、Gamess Input Examplesを補完する目的でも使える。まぁ、計算化学系の研究室にいれば、こういったノウハウとかこつみたいなものは、誰かに聞けばすぐに解決するようなものなんだが、、、、
製薬系って量子化学計算する人って思ったほどは多くないので、D章の計算実践編とF章の目的別対処法は参考になった。
このように、初学者が悩んだりつまずいたりしそうなところをQ&A形式で解説している本なので、実験グループで量子化学計算を担当された方を対象にしているという前書きは納得がいく。
ただし、Howto本ではないから、ザボとか一通り読んであって、Gaussian,GAMESS触ったことがあって、unix系のエディタでファイル操作が普通にできるヒト向けでしょう。あとは、より深く知るためのリファレンスが少なすぎる気がするなぁ。改訂版出すならそこら辺を充実してくれればありがたい。
Wikiあればいいのに、RjpWikiの量子化学計算版みたいなの。
 すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)
すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)
 システムバイオロジーの展開―生物学の新しいアプローチ (Springer reviews)
システムバイオロジーの展開―生物学の新しいアプローチ (Springer reviews) 交響詩篇エウレカセブン DVD-BOX (初回限定生産)
交響詩篇エウレカセブン DVD-BOX (初回限定生産) すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)
すぐできる 量子化学計算ビギナーズマニュアル (KS化学専門書)