13092024 chemoinformatics life
ちょっと間が空いてしまいましたが、MIshima.syk #21をやります。
発表したい方は早めに時間を取っておいてください(いつも駆け込みでぶっこんでくるので、、、) それからオンラインで参加したい場合には私に連絡をください。zoomのアドレスを送ります。
今回はLLMの話とか、ケモインフォマティクスの深い話とかになりそうですが、今回もディープな話で盛り上がれることを期待しています。
余談ですが、今JCUP XIIに出ているのですが、知り合いからラボの先輩がYouTuberになって量子化学に関して熱く語っていという話を聞いて驚きました。
そして私の講演は難しくてわからんとも。わかりやすいように周辺知識も追加したのに、、、 ちなみにトークでの私の主張は以下です。
- Our aim is to describe the ligand association/dissociation process as a chemical reaction, where reactants and products remain unchanged.
- LBDD can be regarded as SBDD in ligand-protein complex is unknown.
共感してくれる人がいると嬉しいのですが。
 計算化学(第3版)
計算化学(第3版)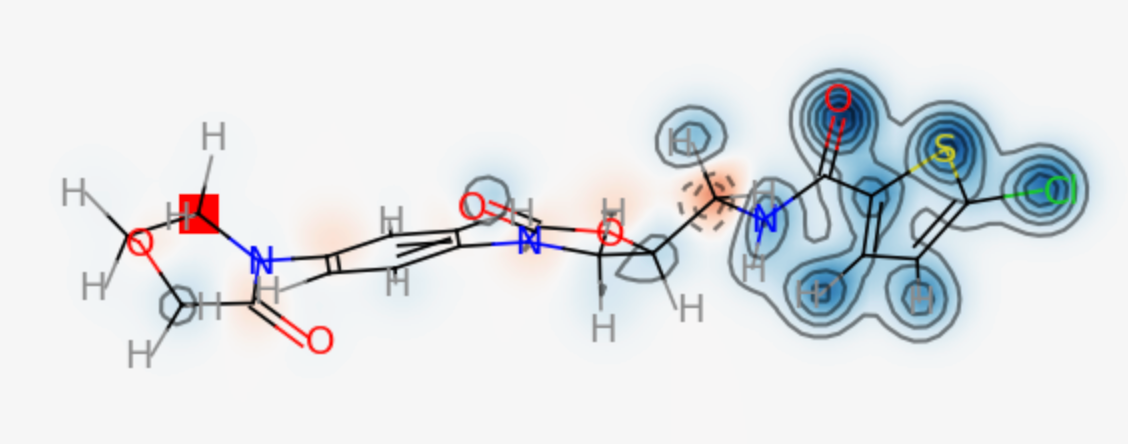 Fig. 1 : F+(r) of Rivaroxaban
Fig. 1 : F+(r) of Rivaroxaban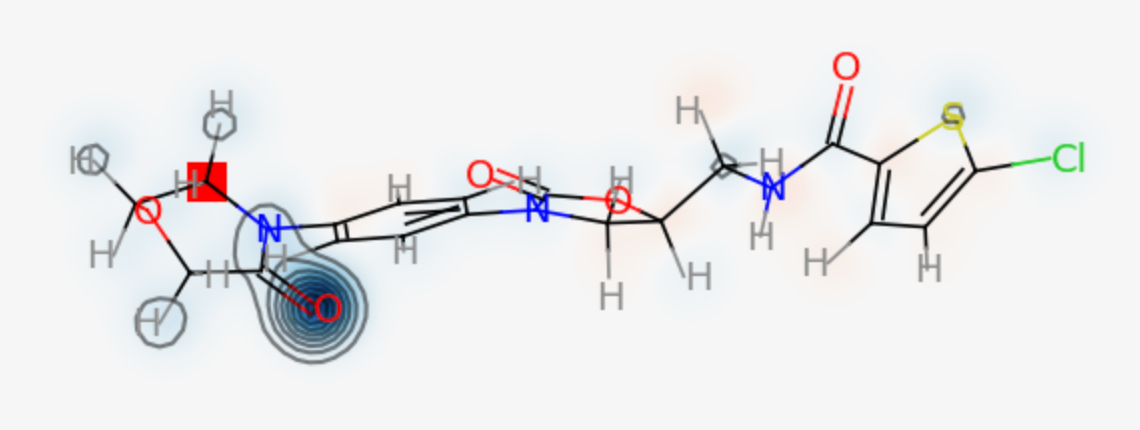 Fig. 2: F-(r) of Rivaroxaban
Fig. 2: F-(r) of Rivaroxaban