
13082022 chemoinformatics FMO SBDD
Evotecから出たこの論文なかなか面白かったですね。"なかなか"というのは我々随分前にこのあたりは通過済みですので。自分たちは取り扱わない蛋白質の事例が多かったので、私にとっては新しく面白い相互作用をいくつも知ることができて満足です。
[ASAP] Hotspot Identification and Drug Design of Protein–Protein Interaction Modulators Using the Fragment Molecular Orbital Method https://t.co/G6H6sGQ0Ha #chemoinformatics #feedly
— kzfm (@fmkz___) August 8, 2022
本論文ではPPIでやっていましたが、ペプチド(環状ペプチド)でも同じことができます。というより我々はそっちを精力的にやっていました。ペプチドは蛋白質と低分子の二重性を示すので面白いモダリティだなぁと思います(課題も多いですがw)このようなFMOの計算結果を用いて、Protein engineeringでより結合の高い蛋白質の設計をしたり、ペプチドの場合より結合能の高いペプチド設計をしたり、それをミミックするような別の分子サイズに置き換えていくのがスタンダードになっていくのだと思います(解釈もしやすいですしね)。
で、こういう仕事を当たり前にやっている状況になると、こんな感じの論文に対し、FMOで論理的に低分子にできるのか(むしろなんでやってないの)?みたいなreplyが脊髄反射でされるようになるわけです。
[ASAP] Peptide-to-Small Molecule: A Pharmacophore-Guided Small Molecule Lead Generation Strategy from High-Affinity Macrocyclic Peptides https://t.co/uoSXodS6HC #medchem #feedly
— kzfm (@fmkz___) August 4, 2022
(鍵垢なので、IDは伏せます)
I wonder if the (inter/intra) FMO interaction patterns with the protein is similar with the peptide and the small ligand
あとはさっさと計算走らせて(大体のpdbファイルは数分でFMO計算走らせられる環境になってる)、暇なときに解析して知識を積み上げるだけのルーチンワークですし、私としてはこういう論文を見つけては、自分で計算したり、チームメンバーのトレーニングとして計算と解釈してもらったりしてますけどなかなか楽しいのでおすすめです。
FMO計算の先について
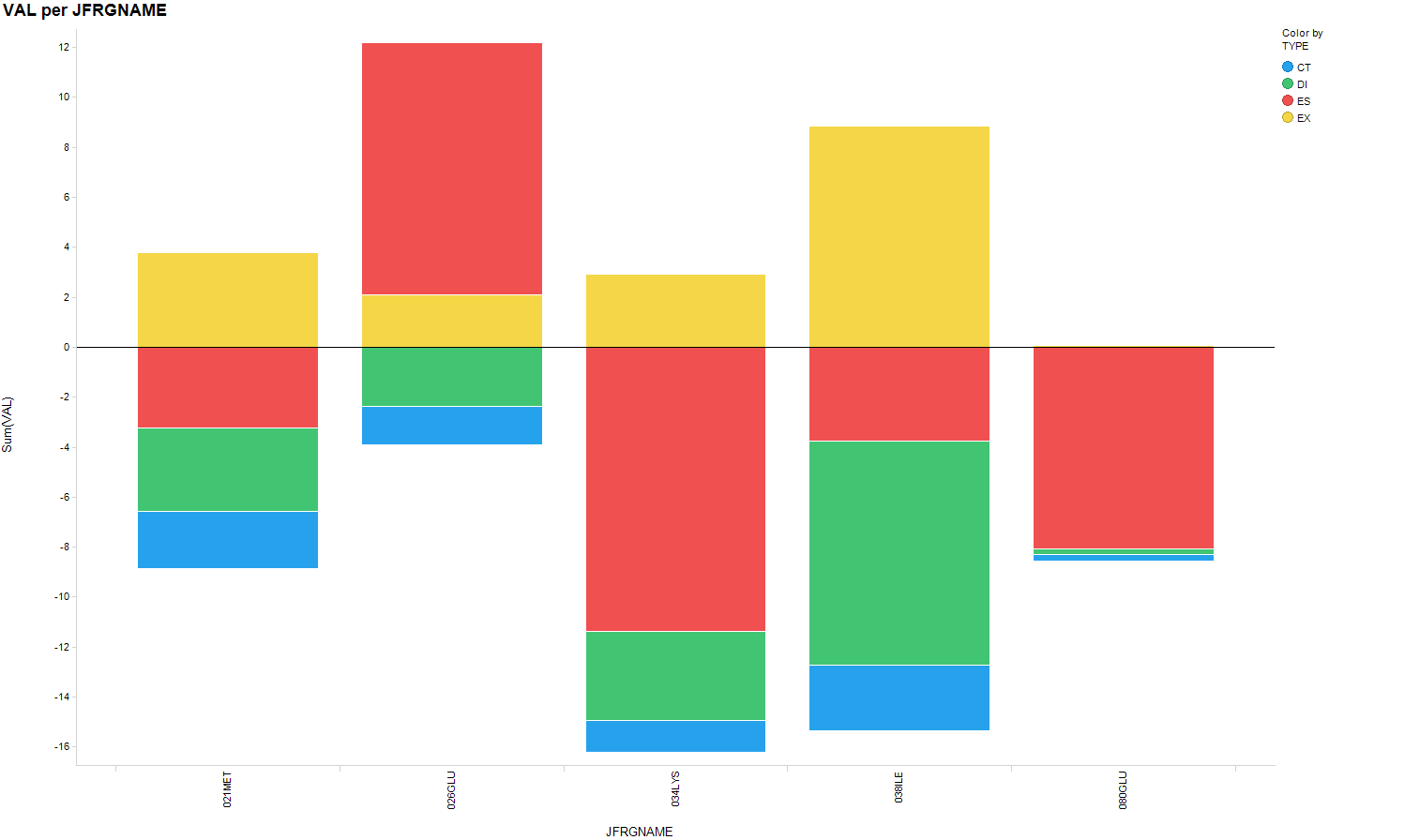
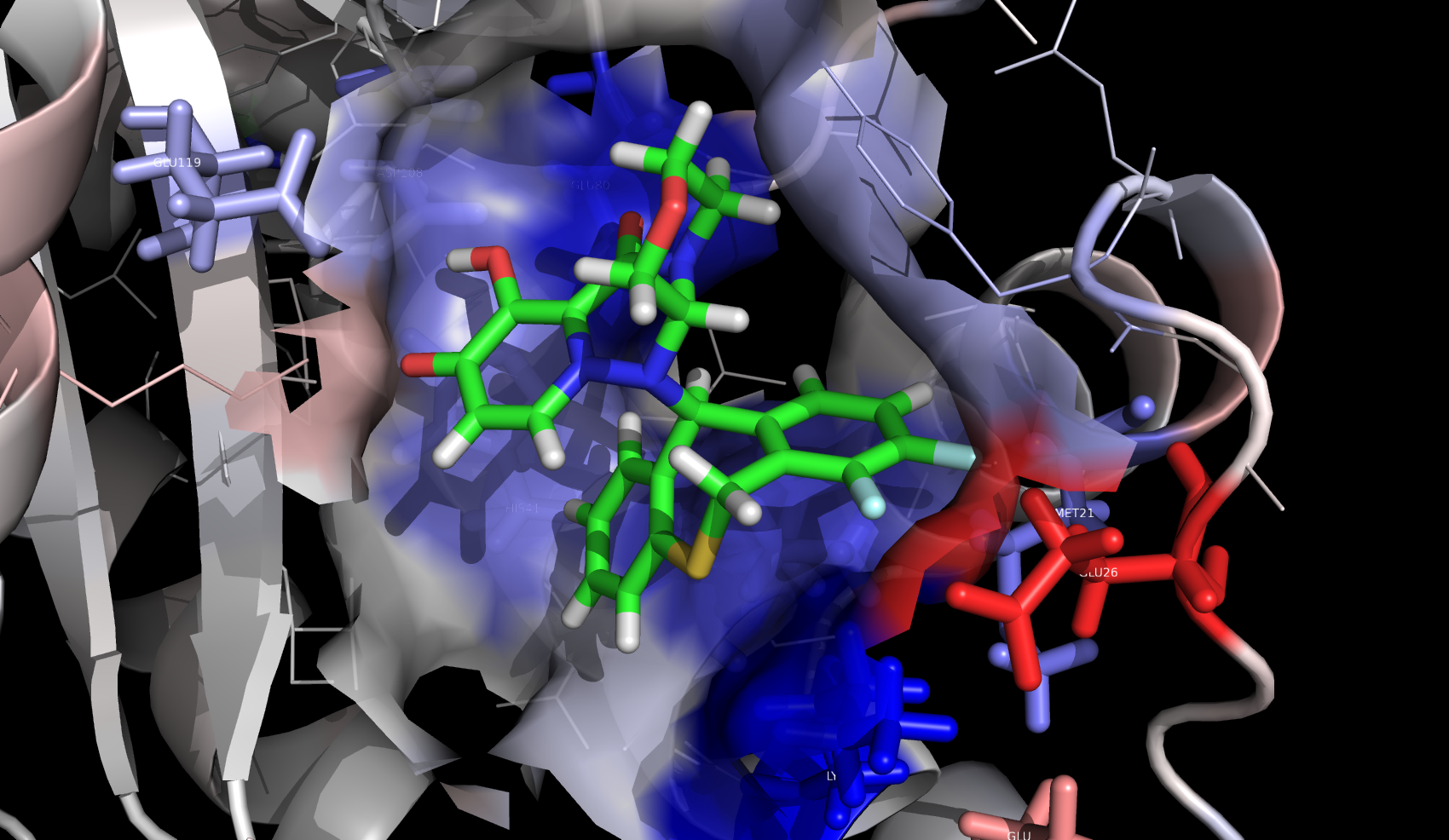
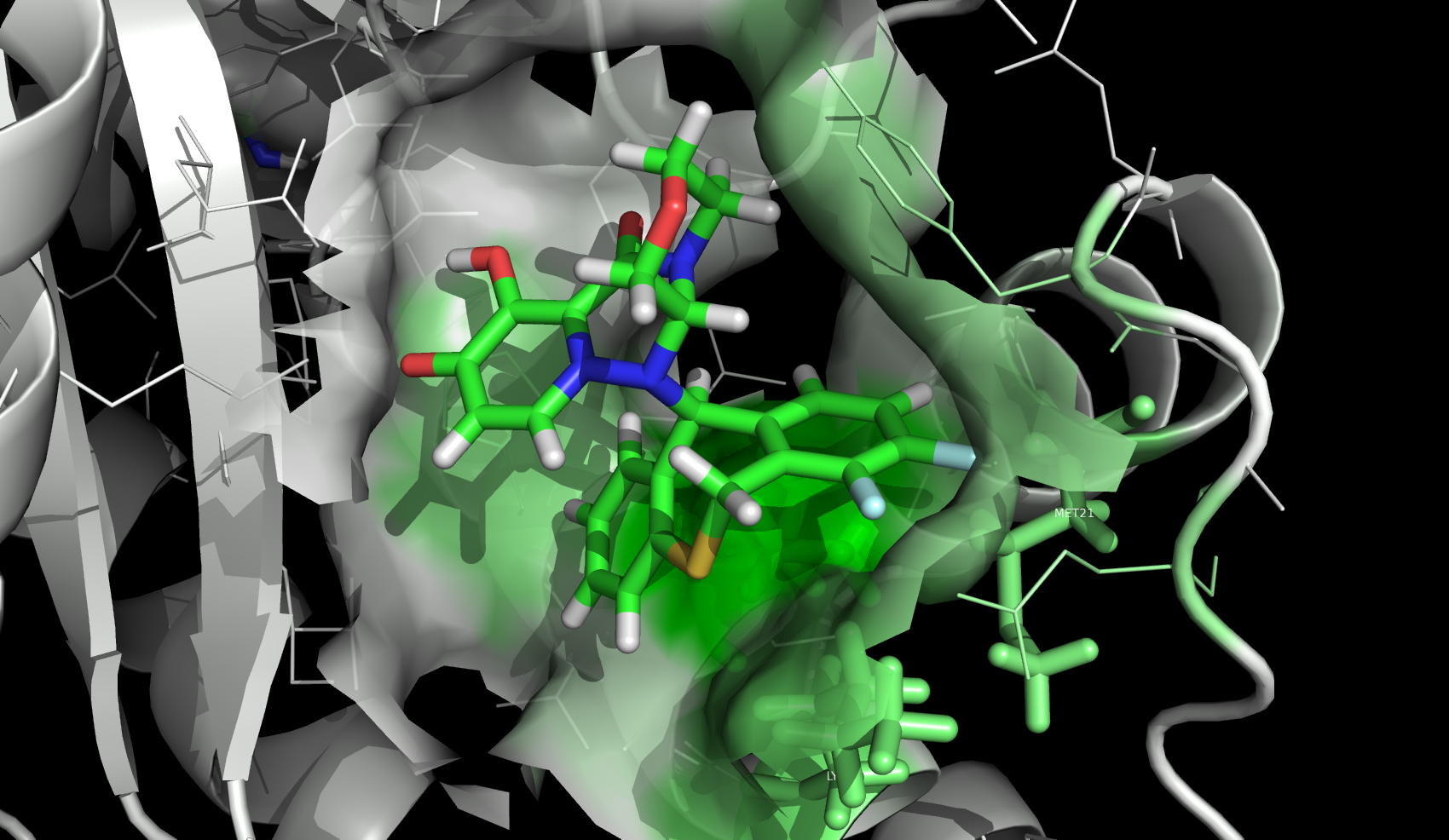
Evotecの論文だとFig. 4.のLIMK1とCofilin1におけるM115とY514の相互作用とか4kcal/molくらいあるし、Fig.6. のKRAS/SOS1のN879のやたら強い相互作用(hotspot)などは明確に見えてきて面白いのだが、IFIEだと相互作用の強さが定量的に出てくるだけだし、PIEDAは成分に分解するだけでDIが効いているのかESが効いているのかどういう成分が関与しているのかがわかるだけでありこのあたりが(いまのところの)FMOの限界ですよねぇと。
この情報をDrug Designに還元するためにはもう少し理解度をあげないといけなくて、どういう軌道の相互作用により強い相互作用が形成されているのかを理解した上で、化合物のこの場所にこういう置換基を導入して軌道の形を変えてやるべきというような論理性が求められてくるのだと思います。
レトロスペクティブな考察を加えるならこの論文も置換基効果で説明できるタイプのものじゃないかなぁと思います。今度計算してみてきれいな結果が出るようだったらブログネタにするかどっかにポスターでも出しに行こうかなぁと思っています。
It’s a tricky PPI that one. https://t.co/u8iusYkwAI
— Darryl B McConnell (@D_B_McConnell) August 9, 2022
実際ノンクラシカルな相互作用がFMOで確認されたら、関与しているフラグメントのHOMO/LUMOチェックしてフロンティア軌道論で説明できるかどうか評価するところまでは基本路線だからね。メドケムの人もリード化合物のHOMO/LUMOくらいは基本として抑えておくと最適化の質が上がるとおもんですけどあんまりやる人いませんよね。 そんなこんなでそういうのPythonでサクッとやりたいよねーといいながらpen先生と一緒に作ったのがpsikitだったりします。
今後のSBDDで重宝されるのは量子化学をきちんと理解してSBDDに活かせる人材かNMR等の実験系をきちんと理解した上でそういう情報とMDを組み合わせてプロジェクトに貢献できる人なんじゃないかなぁと思っています。
FMOについて
FMOは北浦先生により生み出された日本初の手法ですので、Evotecみたいな論文発表が日本からなされると嬉しいですね。FMODDというコンソーシアムもあるので頑張って欲しいです。
うちもなんか貢献を見せないといけませんなぁと思っていますので、近いうちになんか出すと思います。
あとは自分の関与している学会で「ディープすぎるFMO創薬の話」でも企画すればいいんですかねぇ(興味あります?)
 The Fragment Molecular Orbital Method: Practical Applications to Large Molecular Systems
The Fragment Molecular Orbital Method: Practical Applications to Large Molecular Systems ピューと吹く!ジャガー モノクロ版
ピューと吹く!ジャガー モノクロ版