趣旨としては「メディシナルケミストは尊い」となります。
詳細省きますが、最近違いを理解していない人が多すぎてげんなりすることが起こったのと、今週末のCBI講演会でこのあたりの話題が話されるので宣伝がてら書いてみます。
下の図は10年くらい前の某BIソフトウェアのユーザー会で発表したときの資料を少し改変したもので、私がメディシナルケミストをどう定義しているかというものです。
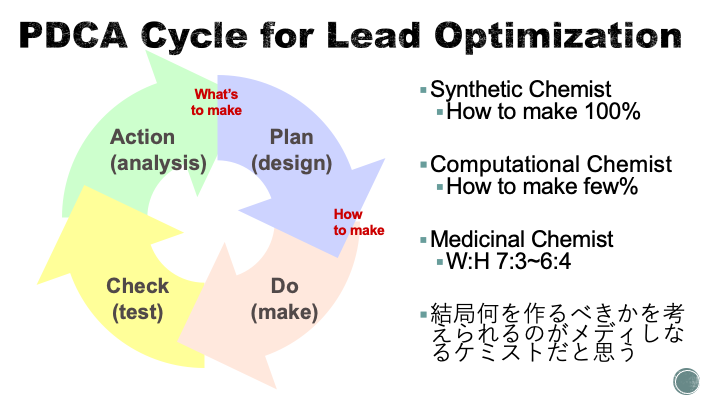
創薬プロジェクトで合成化学者の果たす役割はいわゆる創薬プロジェクトのPDCAサイクル(DMTA)において
- What's to make (何を合成するか)
- How to make (どうやって合成するか)
の2つに大別されるかと思います。典型的な合成化学者とか合成CROは与えられた化合物をどうやって合成するのかを考えることに100%コミットします。一方で計算化学者/Chemoinformatist/SBDDerは分析結果から次に何を作るべきかに全振りします。
メディシナルケミストはWhat's to make:How to makeの割合が7:3-6:4くらいではないかと思われますが、海外最大手のPは10:0に全振りしていると聞いたことがありますというか、前職の上司がそれが「なんか違うわー」ってことで辞めたと聞きました。
10年前の私は上のスライドに書いてあるように「何を作るべきかを考えられるのがメディシナルケミスト」というポジションだったので、将来的には分析できるChemoinformatist/SBDDerが合成CROをうまく使いこなせば十分にメディシナルケミストとなりうるだろうと考えていました。実際REINVENTが出たりDeepLearningによる逆合成が急速に発展したしね。
ただ、実際ケミストをマネージメントする立場になってみると合成化学知らないとかなり困るなぁと(一応合成化学専攻出てるけどw)能力不足を感じたり、やっぱり機械学習は所詮機械学習で、既存の反応の組み合わせしか辿れないというか未知の反応を提案することができないというか、それはAIが将棋で「銅」っていう新しい駒を作り出せないのと一緒なのかなぁと最近は考えを改めています。
というわけで、きちんと合成化学のバックグラウンドを持った人がWhat's to makeのための技術を獲得するというのが真なるメディシナルケミストなのかなぁと思うようになりました。
とはいえ、What's to makeに全振りしてDruglikenessだけきっちり押さえればSynthetic Feasibilityは別に無視して誰かに任せてもいいというような道もあるよなぁとは思います。
