
PerlMolで化学反応のルーチンってどう書けばええの?と聞かれたことはないが、こんな感じで僕は書いてマスヨってことで、ひとつDiels-Alder反応をさせてみる。
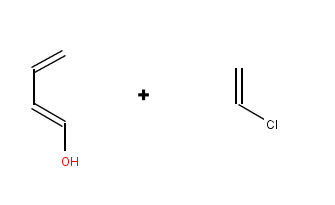
適当に用意したこんな反応だ。
#!/usr/bin/perl
use strict;
use warnings;
use Chemistry::File::SMILES;
use Chemistry::File::SMARTS;
use Chemistry::Ring 'aromatize_mol';
#反応させる基質を用意
my $react1 = Chemistry::Mol->parse('ClC=C dienophile', format => 'smiles');
my $react2 = Chemistry::Mol->parse('OC=C-C=C diene', format => 'smiles');
#dieneとdienophileのパターンをSMARTSで定義
my $dienophile_pat = Chemistry::Pattern->parse("C=C", format => 'smarts');
my $diene_pat = Chemistry::Pattern->parse("C=C-C=C", format => 'smarts');
#まずおまじない
aromatize_mol($react1);
aromatize_mol($react2);
#生成物に名前をつけ,基質をくっつける。
my $name = $react1->name . "+" . $react2->name;
my $prod = Chemistry::Mol->new(name => $name);
$prod->combine($react1, $react2);
#dieneとdienophilieのパターンを探し、みつかったら各オブジェクトにマップ
$dienophile_pat->match($prod);
my @atom_map1 = $dienophile_pat->atom_map;
my @bond_map1 = $dienophile_pat->bond_map;
$diene_pat->match($prod);
my @atom_map2 = $diene_pat->atom_map;
my @bond_map2 = $diene_pat->bond_map;
#反応用の原子が揃っているかチェック
if ($atom_map1[0] && $atom_map1[1] && $atom_map2[0] && $atom_map2[3]) {
#結合の作成
$prod->new_bond(atoms => [$atom_map1[0], $atom_map2[0]], order => '1');
$prod->new_bond(atoms => [$atom_map1[1], $atom_map2[3]], order => '1');
#dieneとdienophileの結合次数を変える
$bond_map1[0]->order(1);
$bond_map2[0]->order(1);
$bond_map2[1]->order(2);
$bond_map2[2]->order(1);
my $smi = $prod->print(format => 'smiles', unique => 1, name => 1);
#SMILESで出力
print "$smi\n";
}
早速実行してみると、
$dareac.pl
OC1C=CCCC1Cl dienophile+diene
jchempaintで確認してみると
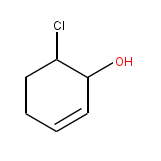
ちゃんと環まいとるヨ。
さて、スクリプトをくどくどと書いたけど、処理の流れは単純でわかりやすいのですヨ。
- 基質の用意と反応する部分構造のパターン定義
- 生成物のオブジェクトを作成し基質を放り込む(combine)
- パターンマッチさせたら、原子と結合に位置のマップ
- マップされた結合と原子に対して処理
- 結合新しく作ったり、切り離したり
- 原子オブジェクトの削除とか
エッジとノードで表現するせいなんだと思うがマップされた結合と原子に対して処理するルーチンが結構煩雑になり気味で、デバッグは大抵ココに集中したりする。dieneの端っこ結合作ったら、pな軌道が隣り合うから間は二重結合になるだろって思うが $bond_map2[1]->order(2)って明示しないと、ラジカルとして表現されちゃったりとか。sp2,sp3の情報入れとけば自動で判別するようにできるような気がするが難しいのだろうか?
あとは、サンプルなのではしょってますが、この反応ではpara体も生成させることは出来ます。
#結合の作成 $prod->new_bond(atoms => [$atom_map1[0], $atom_map2[3]], order => '1'); $prod->new_bond(atoms => [$atom_map1[1], $atom_map2[0]], order => '1');
って書けばpara体出来上がり。
というわけで、普通は二通りの生成物を提示するんだろうけど、
Diels-Alder付加体の二つの置換基はオルト位/パラ位を占めるように位置選択的に付加することが有機電子論から経験的に予測される(オルト-パラ則)。詳細にはFrontier軌道のローブ係数を求めて比較することで説明がなされる。すなわち、下図のようにHOMO/LUMOの係数が大きい点同士が重なるように付加する。
とあるように、軌道の計算してやればどっちが出来やすいか予測することは可能なので、反応処理ルーチンにPyQuanteみたいな量子化学計算モジュールを組み込んで判断させてやれれば素敵かなぁと思っている。